18017847121

AR抑制劑不能增強PARP抑制劑對HR正常前列腺癌的療效,反而通過阻滯細胞周期削弱其作用。DNA修復不受AR直接調(diào)控,協(xié)同作用缺乏理論支持。成果發(fā)表在PNAS雜志(IF:9.1);

《Proceedings of the National Academy of Sciences of the United States of America》簡稱PNAS,創(chuàng)刊于1915年,是美國國家科學院的guan方學術期刊,是shi界gong認的*綜合性科學周刊之一。它致力于發(fā)表具有杰出原創(chuàng)性、重要科學貢獻和廣泛跨學科影響的前沿研究。發(fā)表涵蓋生物科學、物理科學、社會科學、數(shù)學和工程學等幾乎所有自然科學與社會科學分支的研究。年文章數(shù)約3000篇;提供混合開放獲取模式。
當前,針對同源重組修復功能正常的去勢抵抗性前列腺癌,臨床前研究曾提出雄激素受體(AR)通路抑制劑與聚腺苷二磷酸核糖聚合酶(PARP)抑制劑聯(lián)合治療的協(xié)同作用假說。該假說基于AR可調(diào)控DNA損傷修復基因(如BRCA1/2)表達的理論,認為抑制AR通路可誘導“BRCAness"表型,從而增強腫瘤對PARP抑制劑的敏感性。然而,多項臨床研究顯示,該聯(lián)合方案在HR正常患者中僅帶來有限的生存獲益,且美國食品藥品監(jiān)督管理局未批準其用于HR正常人群,凸顯了現(xiàn)有理論與臨床結果之間的顯著矛盾。這一爭議促使研究人員重新審視AR與DNA修復之間的調(diào)控關系,并深入探究在HR正常前列腺癌模型中AR抑制劑與PARP抑制劑相互作用的真實機制。
研究方法:
研究采用多種同源重組修復功能正常(HR-proficient)的前列腺癌細胞系(包括去勢敏感型LNCaP和去勢抵抗型22RV1、LNCaP-abl等)以及HR缺陷型對照細胞(BRCA2缺失的LuCaP176細胞),系統(tǒng)評估了雄激素受體靶向療法與PARP抑制劑的相互作用。在方法學上,研究通過細胞活力檢測(Cell Titer Glo)評估藥物單用及聯(lián)用的效果;利用γH2AX和RAD51免疫熒光染色定量分析DNA損傷與修復狀態(tài);采用RNA測序和ATAC測序分別在轉錄組和染色質(zhì)可及性層面解析藥物處理的分子變化;并通過免疫印跡驗證關鍵蛋白表達。為驗證細胞周期的作用,研究使用了CDK4/6抑制劑帕博西尼(palbociclib)和CDK7抑制劑SY-1365進行功能干預。此外,研究還結合了公共臨床轉錄組數(shù)據(jù),分析雄激素剝奪治療(ADT)前后患者腫瘤中細胞周期相關通路的變化,從而在臨床樣本層面佐證機制發(fā)現(xiàn)。整體實驗設計涵蓋了從體外模型到臨床關聯(lián)的多層次驗證,系統(tǒng)闡明了藥物響應機制。
1. AR抑制在前列腺癌去勢敏感或去勢抵抗模型中均不與PARP抑制產(chǎn)生協(xié)同作用
在雄激素剝奪或使用恩雜魯胺抑制AR信號后,去勢敏感型前列腺癌細胞(LNCaP)對多種PARP抑制劑(包括奧拉帕利、盧卡帕利、他拉唑帕利及AZD-5305)的敏感性顯著下降,表現(xiàn)為IC50值上升。然而,在去勢抵抗型前列腺癌細胞系(22RV1、LN95、LNCaP-abl)中,AR抑制并未改變其對PARP抑制劑的敏感性。即使在臨床相關濃度下(10 μM恩雜魯胺與1 μM PARPi),聯(lián)合用藥也未能比單一用藥顯示出額外的細胞生長抑制優(yōu)勢。這些結果證明,AR抑制不僅無法與PARP抑制劑在HR正常的癌細胞中產(chǎn)生協(xié)同作用,反而在去勢敏感模型中會削弱PARP抑制劑的療效。

圖1,AR抑制不改變?nèi)莸挚剐郧傲邢侔?/span>PARP抑制的敏感性
2. PARP抑制劑治療可調(diào)控AR相關基因的表達
隨后,研究人員通過轉錄組分析揭示了PARP抑制劑在有無AR抑制條件下對HR正常前列腺癌細胞基因表達的影響。結果顯示,PARP抑制劑處理顯著誘導了CYP家族基因(如CYP1A、CYP1B)的表達。通路富集分析表明,在雄激素(DHT)存在的條件下,PARP抑制劑激活了p53通路并誘導細胞凋亡,同時抑制了細胞周期相關通路(如E2F靶標、G2/M檢查點);而在雄激素剝奪條件下,這些細胞周期和凋亡通路的改變顯著減弱甚至消失。這一發(fā)現(xiàn)與圖1中觀察到的生長抑制結果一致,表明PARP抑制劑的抗癌效果依賴于雄激素存在下的細胞周期進程,而非通過AR直接調(diào)控的DNA修復通路。

圖2,PARP抑制誘導細胞周期和DNA損傷應答通路的變化
進一步的研究結果揭示了PARP抑制劑對AR驅動轉錄的調(diào)控作用及機制。RNA-seq分析顯示,在DHT刺激的22RV1細胞中,PARP抑制劑能夠抑制一部分雄激素響應基因的誘導表達(Cluster 2與Cluster 4),這些基因富集于細胞周期與代謝通路;而在LNCaP細胞中,PARP抑制劑僅對少量AR調(diào)控基因有抑制作用。ATAC-seq分析進一步發(fā)現(xiàn),PARP抑制劑(如奧拉帕利)在DHT存在時會導致一部分染色質(zhì)區(qū)域的可及性降低,但這種變化與基因表達改變的重合度很低。例如,LRRC4基因的可及性雖受DHT調(diào)控,卻不受奧拉帕利影響。這些結果綜合表明,PARP抑制劑對AR調(diào)控基因表達的影響主要在轉錄水平發(fā)生,而非通過改變?nèi)旧|(zhì)可及性來實現(xiàn)。

圖3,PARP抑制劑對AR驅動轉錄的調(diào)控作用及機制
3. 雄激素剝奪或AR抑制不會抑制DNA修復。
RNA-seq分析顯示,無論是在雄激素剝奪還是DHT處理的條件下,DNA修復相關基因(包括先前報道的AR靶基因如BRCA2)的表達均未受到顯著抑制。ATAC-seq數(shù)據(jù)進一步證實,AR抑制并未改變BRCA1/BRCA2等關鍵修復基因位點的染色質(zhì)可及性。功能實驗通過γH2AX焦點形成檢測發(fā)現(xiàn),在去勢敏感LNCaP細胞中,雄激素剝奪反而減弱了PARP抑制劑(奧拉帕利)誘導的DNA損傷;而在去勢抵抗22RV1細胞中,DNA損傷水平不受雄激素狀態(tài)影響。這些證據(jù)一致表明,AR通路抑制并不直接削弱前列腺癌細胞的DNA修復能力,也無法增強PARP抑制劑所致的DNA損傷。

圖4,AR不直接調(diào)控DNA修復基因的表達
4. 細胞周期進展是響應PARP抑制的必要條件
研究發(fā)現(xiàn),在去勢敏感LNCaP細胞中,高劑量DHT(10 nM)誘導的細胞生長抑制會嚴重削弱奧拉帕利的療效,其效果甚至強于雄激素剝奪。為驗證這一現(xiàn)象是否存在于患者體內(nèi),研究人員分析了公共臨床測序數(shù)據(jù),發(fā)現(xiàn)雄激素剝奪治療(ADT)顯著抑制了患者腫瘤中細胞周期相關通路(E2F靶點、G2/M檢查點)的基因表達,確認ADT在臨床上確實會引起細胞周期停滯。為直接驗證細胞周期的作用,研究使用CDK4/6抑制劑帕博西尼(Palbociclib)誘導G1期阻滯,發(fā)現(xiàn)這顯著降低了多種PARP抑制劑(奧拉帕利、盧卡帕利、他拉唑帕利、AZD-5305)在HR正常前列腺癌細胞中的療效。這種效應并不僅限于PARP抑制劑,同樣也削弱了傳統(tǒng)DNA損傷化療藥物(如順鉑、依托泊苷、多柔比星)的作用,表明細胞周期依賴性是DNA損傷類藥物的普遍機制。有趣的是,在HR缺陷(BRCA2缺失)的LuCaP176細胞模型中,使用CDK7抑制劑SY-1365抑制細胞增殖,并未削弱奧拉帕利的療效。這揭示了一個關鍵差異:細胞周期進展僅是HR正常細胞響應PARP抑制的必要條件,而在HR缺陷細胞中并非必需。
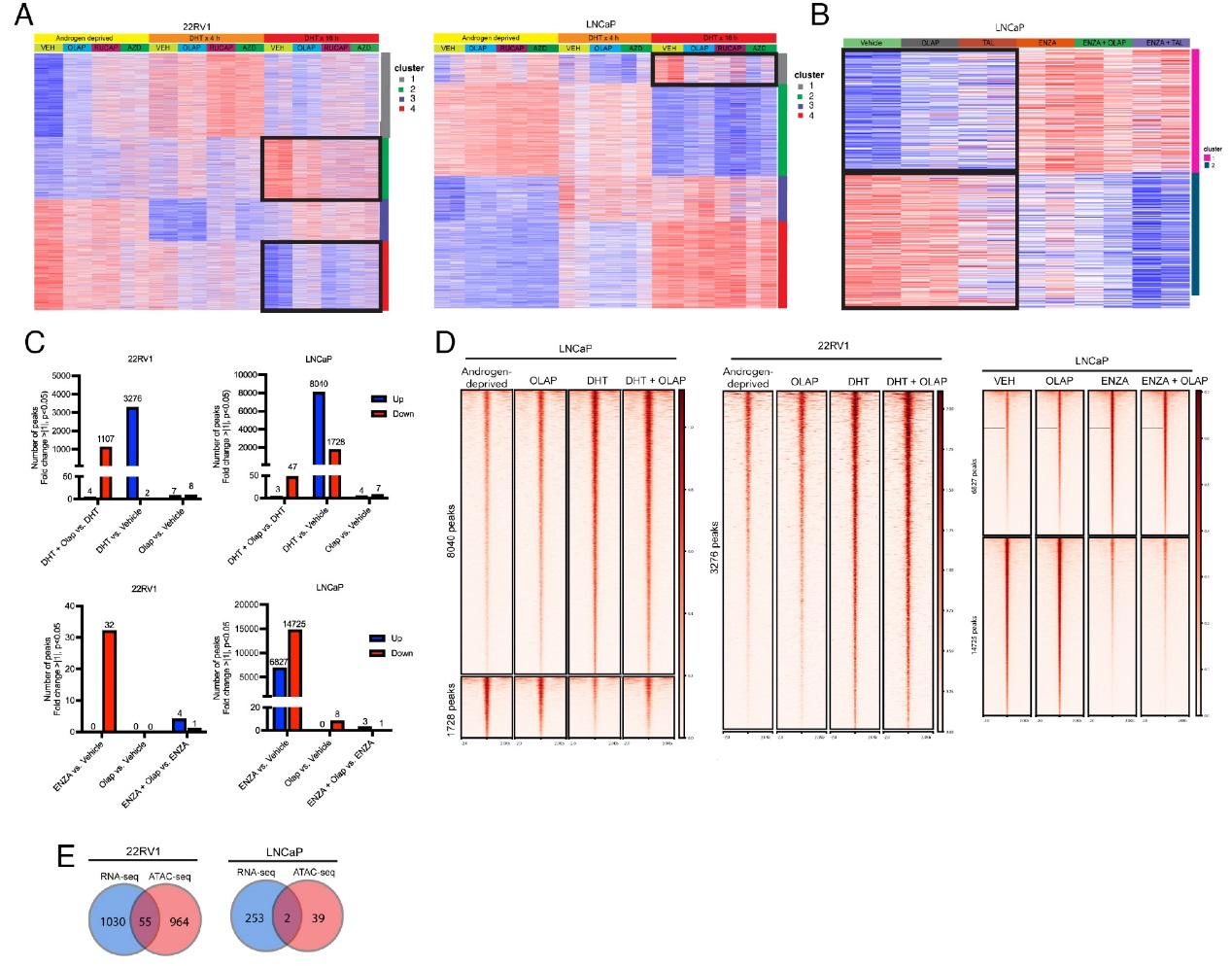
圖5,抑制細胞周期進展會削弱對PARP抑制劑的治療反應。
5. 全文總結
本研究dian覆了“AR抑制劑通過抑制DNA修復來協(xié)同PARP抑制劑"的傳統(tǒng)理論,shou次揭示在HR正常前列腺癌中,細胞周期進展才是PARP抑制劑起效的關鍵前提。AR抑制劑會誘導G1期阻滯,從而削弱PARP抑制劑的療效,這解釋了二者聯(lián)合療法在HR正常患者中效果不佳的原因。該發(fā)現(xiàn)提示臨床應避免對HR正常患者聯(lián)合使用PARP抑制劑與細胞周期阻滯類藥物(如ADT),同時支持繼續(xù)探索該聯(lián)合方案在HR缺陷患者中的應用。盡管研究主要基于細胞模型且深層機制有待闡明,但其結論為前列腺癌精準治療提供了重要的理論轉向與臨床指導依據(jù)。
 聯(lián)系人:張
聯(lián)系人:張 地址:上海市長江南路180號長江軟件園B區(qū)B637室
地址:上海市長江南路180號長江軟件園B區(qū)B637室 郵箱:2844970554@qq.com
郵箱:2844970554@qq.com 傳真:
傳真:歡迎您關注我們的微信公眾號了解更多信息
 掃一掃
掃一掃 歡迎來到
歡迎來到